
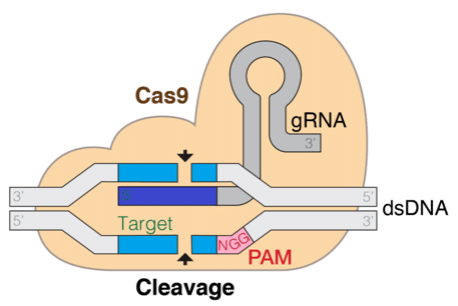
The Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) technology promises to revolutionize genetic editing technologies, which is transformative for understanding and treating diseases. This technique is based on a natural mechanism found in bacteria that allows a protein, coupled with a guide RNA (gRNA) chain, to locate and make specific cuts in the target genome. Being able to computationally predict the efficiency and specificity of the gRNA is crucial for the success of genetic editing.
RNA, transcribed from DNA sequences, is an important type of biological sequence made up of ribonucleotide bases (A, U, G, C), which folds into a three-dimensional structure. Leveraging recent advancements in large-scale language models (LLMs), a variety of tasks in computational biology can be tackled by fine-tuning pre-trained biological LLM models on billions of known biological sequences. Downstream tasks in RNAs are relatively understudied.
In this research, we adopt a pre-trained genomic LLM for gRNA efficiency prediction. The idea is to treat a computer-designed gRNA as a sentence and fine-tune the LLM to perform sentence-level regression tasks analogous to sentiment analysis. We use efficient parameter fine-tuning methods to reduce the number of parameters and GPU utilization for this task.
Large language models (LLMs) have garnered significant interest for their ability to encode syntax and semantics of natural languages. The neural architecture behind LLMs is transformers, consisting of attention-based encoder-decoder blocks that generate an internal representation of the data they are trained on (encoder) and are capable of generating sequences in the same latent space resembling the original data (decoder). Due to their success in natural language, recent works have explored the use of LLMs for molecular biology information, which is inherently sequential.
DNABERT is a pre-trained transformer model with non-overlapping human DNA sequence data. The backbone is a BERT architecture comprised of 12 encoding layers. The authors of this model report that DNABERT is able to capture a good representation of human genome features, allowing cutting-edge performance in downstream tasks such as promoter prediction and splice site identification. We decided to use this model as the basis for our experiments.
Despite the success and popular adoption of LLMs, fine-tuning these models can be challenging due to the number of parameters and computational requirements. For this reason, efficient Parameter Efficient Fine-Tuning (PEFT) methods have been developed. In this research, we utilize one of these methods, called LoRA (Low-Rank Adaptation). We present the method in the following sections.
The goal of this solution is to fine-tune the base DNABERT model to predict the activity efficiency of different gRNA candidates. Therefore, our solution first takes gRNA data and processes it, as described later. Then, we use an Amazon SageMaker notebook and the Hugging Face PEFT library to fine-tune the DNABERT model with processed RNA data. The label we want to predict is the efficiency score as calculated in experimental conditions testing with real RNA sequences in cell cultures. These scores describe a balance between being able to edit the genome and not damaging non-targeted DNA.
For this solution, access to the following items is needed:
– A SageMaker notebook instance (we trained the model on a ml.g4dn.8xlarge instance with a single NVIDIA T4 GPU)
– transformers-4.34.1
– peft-0.5.0
– DNABERT 6
We use gRNA data released by researchers in a paper on gRNA prediction using deep learning. This dataset contains efficiency scores calculated for different gRNAs.
To train the model, a 30-mer gRNA sequence and an efficiency score are needed. A k-mer is a contiguous sequence of k nucleotide bases extracted from a longer DNA or RNA sequence. For example, if one has the DNA sequence “ATCGATCG” and chooses k = 3, the k-mers within this sequence would be “ATC,” “TCG,” “CGA,” “GAT,” and “ATC.”
To encode the gRNA sequence, we utilize the DNABERT encoder. DNABERT was previously trained on human genomic data, making it a good model for encoding gRNA sequences. DNABERT tokenizes the nucleotide sequence into overlapping k-mers, and each k-mer serves as a word in the DNABERT model’s vocabulary. The gRNA sequence is split into a sequence of k-mers, and then each k-mer is replaced with an embedding for the k-mer in the input layer. Otherwise, the DNABERT architecture is similar to BERT. After encoding the gRNA, we use the [CLS] token representation as the final encoding of the gRNA sequence. To predict efficiency scores, we use an additional regression layer. MSE will be the training objective.
Adjusting all parameters of a model is costly because the pre-trained model becomes much larger. LoRA is an innovative technique developed to address the challenge of fine-tuning extremely large language models. LoRA offers a solution by suggesting that the weights of the pre-trained model remain fixed while introducing trainable layers (called low-rank decomposition matrices) within each transformer block. This approach significantly reduces the number of parameters that need to be trained and decreases GPU memory requirements, as most model weights do not require gradient computations.
Therefore, we adopt LoRA as a PEFT method in the DNABERT model. LoRA is implemented in the Hugging Face PEFT library. When using PEFT to train a model with LoRA, the hyperparameters of the low-rank adaptation process and how to wrap base transformer models can be defined.
RMSE, MSE, and MAE were used as evaluation metrics, tested with ranks of 8 and 16. Additionally, a simple fine-tuning method was implemented, which simply adds several dense layers after the DNABERT embeddings. The following table summarizes the results:
| Method | RMSE | MSE | MAE |
|———————|——–|———-|——-|
| LoRA (rank = 8) | 11.933 | 142.397 | 7.014 |
| LoRA (rank = 16) | 13.039 | 170.010 | 7.157 |
| One dense layer | 15.435 | 238.265 | 9.351 |
| Three dense layers | 15.435 | 238.241 | 9.505 |
| CRISPRon | 11.788 | 138.971 | 7.134 |
When the rank is 8, there are 296,450 trainable parameters, which is approximately 33% of the total. Performance metrics are ‘rmse’: 11.933, ‘mse’: 142.397, ‘mae’: 7.014.
When the rank is 16, there are 591,362 trainable parameters, which is approximately 66% of the total. Performance metrics are ‘rmse’: 13.039, ‘mse’: 170.010, ‘mae’: 7.157, indicating a potential overfitting issue under this configuration.
Finally, we compare with the existing method CRISPRon, which is a CNN-based deep learning model. Performance metrics are ‘rmse’: 11.788, ‘mse’: 138.971, ‘mae’: 7.134.
As expected, LoRA works much better than simply adding some dense layers. Although LoRA’s performance is slightly worse than that of CRISPRon, with a thorough hyperparameter search, it is likely to surpass CRISPRon.
We highly recommend shutting down the SageMaker instance when not in active use to avoid unnecessary costs on unused computation.
In conclusion, we demonstrate how to use PEFT methods to fine-tune DNA language models using SageMaker, focusing on predicting the efficiency of CRISPR-Cas9 RNA sequences for their impact on current genetic editing technologies. We also provide code that can help get started with biology applications on AWS.
via: MiMub in Spanish









